

Compare PDB and AlphaFold Structures in iCn3D
The objective of this exercise is to compare an experimentailly determined structure PDB 3NOS, Human Endothelial Nitric Oxide Synthase, with an AlphaFold predicted structure P20474 with NCBI's iCn3D. We will align the structures, assess RMSD and pLDDT, and discuss primary literature deposited with PDB 3NOS. The embedded links labled "follow" are designed to help participants in the live session follow the instructor's directions. If you fall behind or want to view an instructor created rendering, click these "follow" links.
iCn3D Help Docs
1. Go to iCn3D
2. Input your PDB and AlphaFold IDs, "3NOS,P29474", and Click on Load Biological Unit
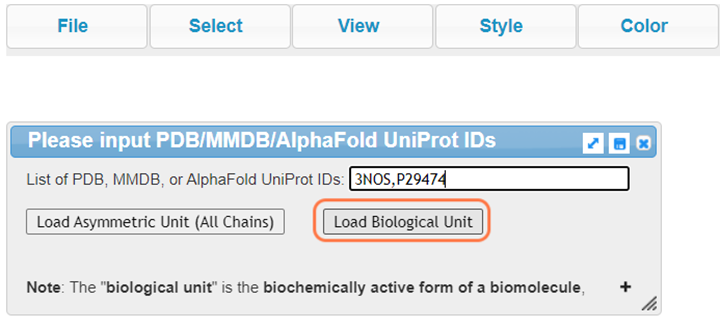
Follow
3. Orient yourself to the structures
Change style, color, and orientation to your liking
4. Realign Structure

5. Select 3NOS_A +Ctrl P29474_A

6. Select 3NOS_A + Alt + P29474_A and click on Realign with TM-align
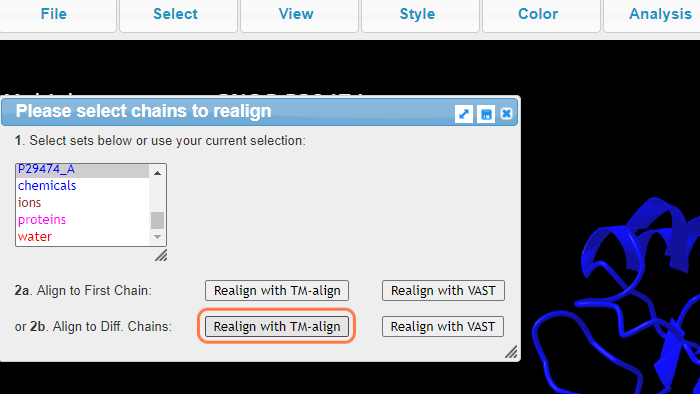
Follow
The aligned section is automatically saved for you as protein aligned in Analysis > Defined sets
7. View alignment

The Root mean square deviation reported of 0.72 is the measure of deviance between the predicted structure and the experimental structure (C alpha atoms only). This is a relatively low RMSD.
The Template modeling score (TM-Score) ranges between 0 and 1, where 1 indicates a perfect match between two structures and 0 indicates no match. Scores of 0.2 are generally considered random, where 0.5 assume a similar fold.
8. Change Color to "AlphaFold Confidence"

Follow
AlphaFold generates a per-residue estimate of confidence in the structure from a scale of 0-100 called pLDDT
-
Ranges at or above 90 are expected to be high accuracy and can be used for atomistic experiments
-
Ranges between 70-90 are pretty good, especially in the backbone
-
Ranges between 50-70 are low confidence
-
And ranges below 50 really shouldn’t be interpreted
9. View full structure by turning off "Selection"

10. Click on View Selection for best view of the alignment

11. View Structure and color-coordinated sequence
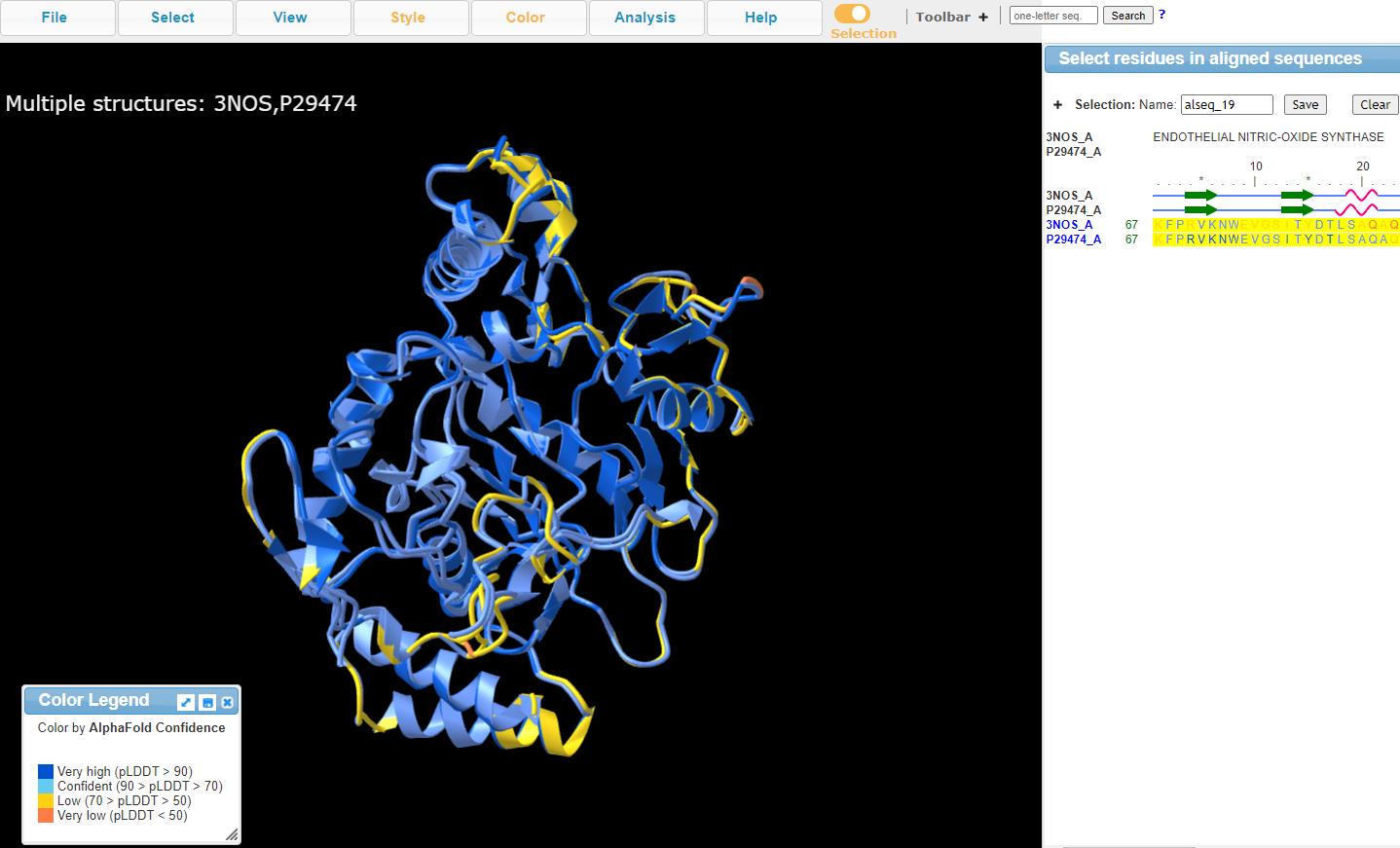
Conclusion:
What do these differences mean? Areas with lower pLDDT could indicate a mistake in the prediction but it could also indicate a mistake in the x-ray structure. Experimental determination methods aren’t perfect and the pairing of these computational and experimental methods can be useful in improving the determination of structures. This specific crystal structure (PDB: 3NOS) has a resolution of 2.4 angstroms. It is possible to misfit the density at that resolution.
Many researchers are starting to compare AlphaFold predicted and solved structures in a fashion similar to this example to identify mistakes in experimental structures.
Last Reviewed: February 21, 2023