

Comparative Genome Viewer - Introduction
- Share





Agenda
- Overview of CGV
- Demonstration of features
- Explore on your own
Overview
Why use CGV?
At a high level, it is good for generating hypotheses, that is, finding interesting patterns and differences that can be investigated further experimentally.
For example, you might want to see how a disease-associated region or gene has evolved in related species.
For example, you might want to see how a disease-associated region or gene has evolved in related species.
More specifically, use CGV to find:
-
- chromosome rearrangements, such as inversions
- large-scale insertions or deletions
- blocks of synteny, such as gene order conservation, between different species
- gene duplications or gene loss
- genomic changes in older and newer assemblies for the same species, or between different strains of the same species
What can CGV do?
-
-
Compare two eukaryotic assemblies
CGV includes animals, plants, and fungi- > 400 assemblies
- ~200 species
- ~480 alignments
-
And continues to grow!
CGV does not include mitchondrial genomes.
-
-
- Different assemblies for the same organism
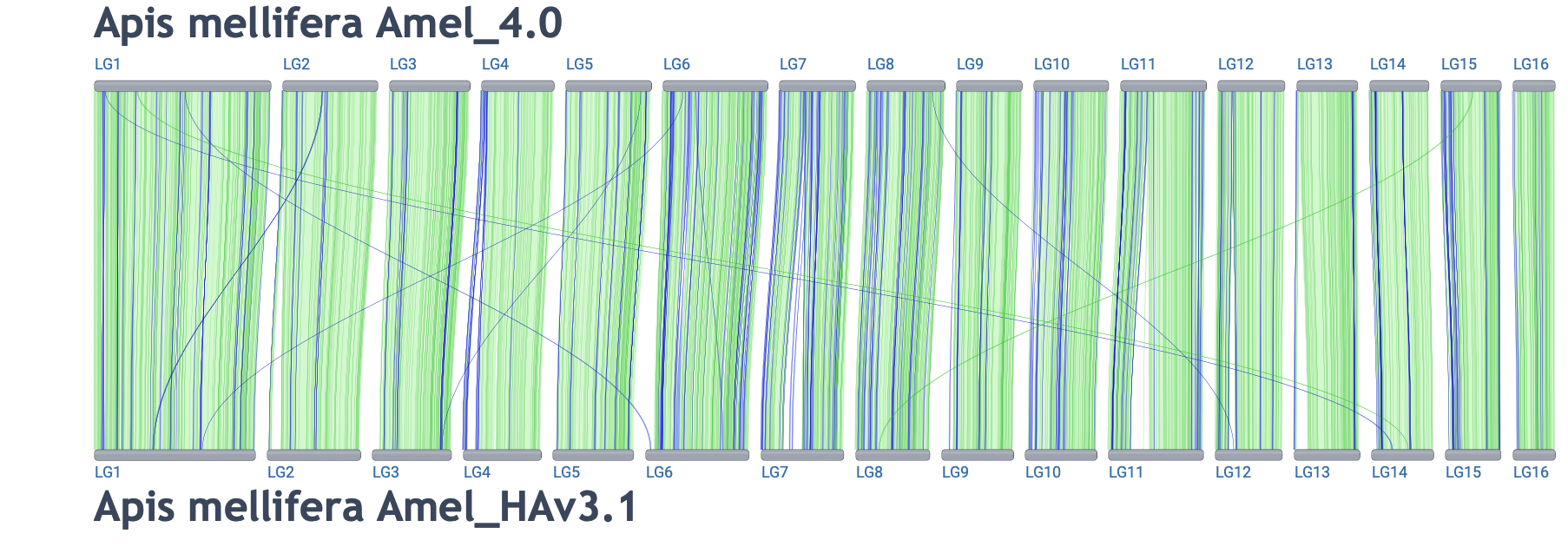
-
-
-
-
- Cross-species comparisons
-
-
-
-

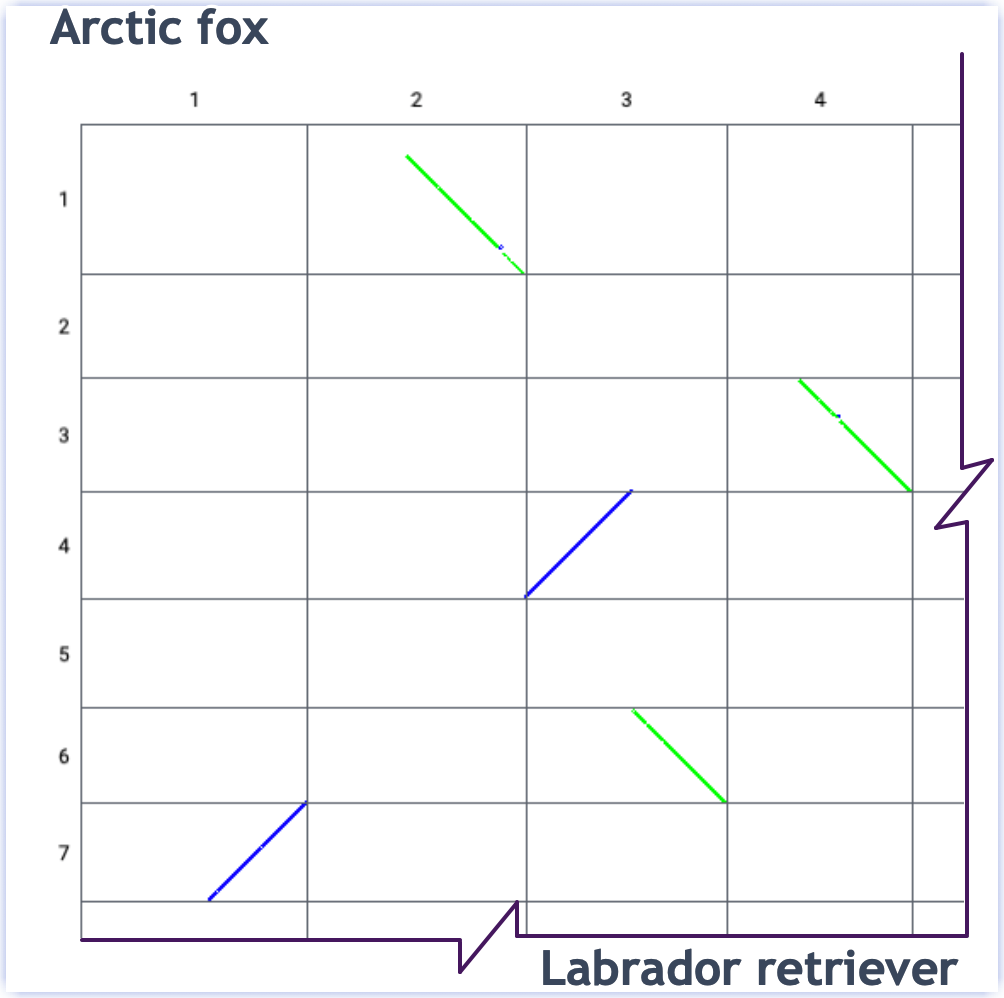
- View as a dotplot
-
-
-
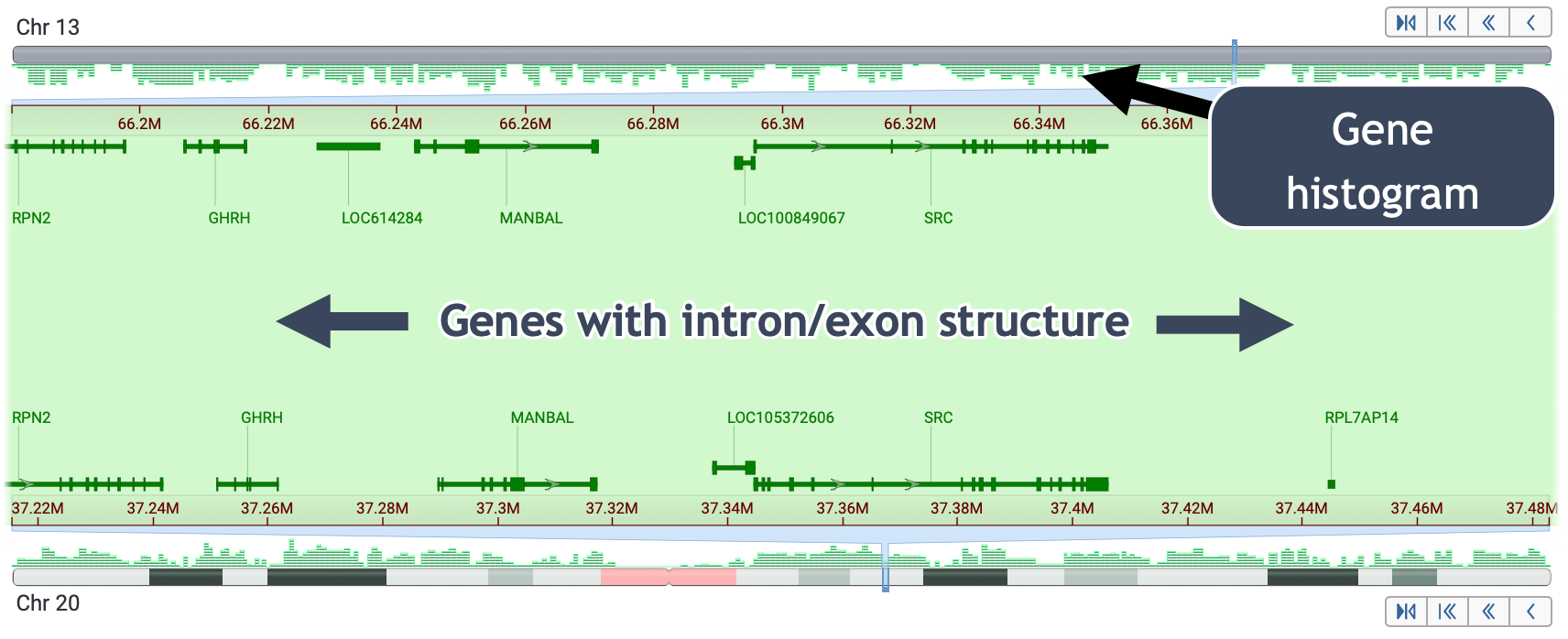
- View chromosomes → genes → sequence
- View at the gene level
-
-
-
- View and analyze at the sequence level four ways
- (This is an image of the pop-up you get when you right-click on an alignment)

- Download a graphics file of the current view
- Download alignment files
- Request alignments
- Help documentation
- For the meaning of "Best reciprocal match", see the link, assembly alignment documentation.
- This is a nice graphic in the Help document:

-
Page 1 of 1
Last Reviewed: July 12, 2023